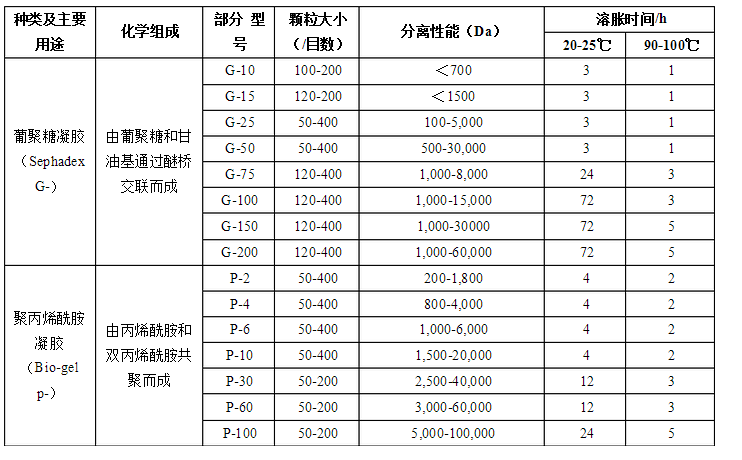
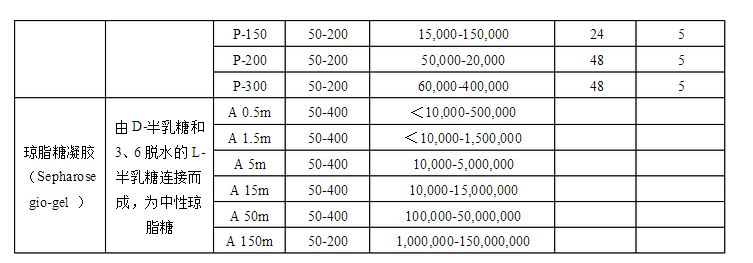
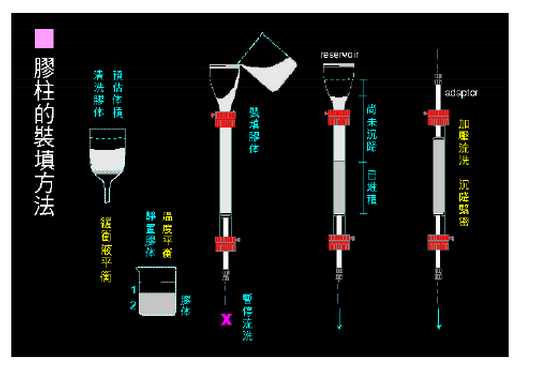
→流動平衡(裝填后放置一段時間,再開始流動平衡,流速應低于層析時所需的流速。在平衡過程中逐漸增加到層析的流速,千萬不能超過**終流速。平衡凝膠床過夜)→落柱頭(小體積,流速壓力判斷,大體積,可接壓力表)
實驗室用:肉眼觀察均勻、無紋路、無氣泡,有色物質如藍色葡聚糖-2000(2mg/mL)、血紅蛋白等上柱,觀察有色區帶在柱中的洗脫行為以檢測凝膠柱的均勻程度。如果色帶狹窄、平整、均勻下降,則表明柱中的凝膠填裝情況較好,可以使用;如果色帶彌散、歪曲,則需重新裝柱。 生產中:用測量層析柱柱效及對稱性的來檢定層析柱是否可投入生產中使用。 注:新凝膠柱可以能過提高緩沖體系中鹽離子的濃度來降低非特異性吸附的程度。 層析柱的處理:
加樣前去除樣品中的不溶物,樣品濃度與分配系數無關,查粘度影響運動。 加樣要求快速均勻,加樣體積,分級分離時為柱體積的1%~5%,分組分離時為柱體積的10%~30%。 洗脫:
半收縮保存:用完以水沖洗,60%~70%酒精沖洗,待凝膠體積縮小,浸泡于70%乙醇中保存; 干燥保存(長期不用):用完后水沖洗,濾干,低濃度乙醇水洗,逐漸加大乙醇用量(50%、70%、90%、95%,防止結塊),**后用95%乙醇洗,全部去水,再用乙烯或乙醚除乙醇,抽濾干,60℃~80℃干燥后保存。
3. 應用:
脫鹽:適用的凝膠為SephadexG-10、15、25及Bio-Gel-p-2、4、6或Ultragel AcA 202等排阻極限較小的凝膠類型 ,柱長與直徑之比為5-15,樣品體積可達柱床體積的25%-30%;(為了防止蛋白質脫鹽
后溶解度降低會形成沉淀吸附于柱上,一般用醋酸銨等揮發性鹽類緩沖液使層析柱平衡,然后加入樣品,再用同樣緩沖液洗脫,收集的洗脫液用冷凍干燥法除去揮發性鹽類。)
溶液濃縮:利用凝膠顆粒的吸水性,將干燥的Sephadex(粗顆粒)加入溶液中,Sephadex可以吸收大量的水,溶液中的小分子物質也會滲透進入凝膠孔穴內部,而大分子物質則被排阻在外,通過離心或過濾去除凝膠顆粒,即可得到濃縮的樣品溶液(這種濃縮方法基本不改變溶液的離子強度和pH值);
分子量的計算與蛋白復性……
離子交換層析(Ion Exchange Chromatography, IEC)
1. 離子交換劑組成:高分子聚合物基質、電荷基團、平衡離子。
親水性:主要分離無機離子、有機酸、核苷等小分子物質;其次用于從蛋白質溶液中除表面活性劑、兩性電解質等;也可用于分離某些不易變性的蛋白質。
陽離子交換劑:電荷基因帶負電,平衡離子帶正電,可與帶正電離子基團發生交換。一般結合磺酸基團(-SO3H),如磺酸甲基(簡寫為SM)、磺酸乙基(SE)等為強酸型離子交換劑;結合磷酸基團(-PO3H2)和亞磷酸基團(-PO2H)為中等酸型離子交換劑;結合酚羥基(-OH )或羧基(-COOH),如羧甲基(CM)為弱酸型離子交換劑。
強酸性陽離子交換樹脂:R-SO3H(磺酸基)和R-CH2SO3H(次甲基磺酸基)R代表基質(樹脂) 中酸性陽離子交換樹脂:R-PO3H2,R-POH2
弱酸性陽離子交換樹脂:R-COOH,R-OCH2COOH,R-C6H5OH等弱酸性基團; 強酸性:R-SO3 -H+
+ Na+
—— R-SO3- Na+
+ H+
弱酸性:R-COOH+Na+
—— R-COONa +H+
陰離子交換劑:電荷基因帶正電,平衡離子帶負電,可與帶負電離子基團發生交換。一般結合季胺基團(-N(CH3)3),如季胺乙基(QAE)為強堿型離子交換劑;結合叔胺(-N(CH3)2)、仲胺(-NHCH3)、伯胺(-NH2)等為中等或弱堿型離子交換劑;如結合二乙基氨基乙基(DEAE)為弱堿型離子交換劑。
強堿性陰離子交換樹脂:活性基團為季胺基團,如三甲胺基或二甲基-ß-羥基乙基胺基 中堿性陰離子交換樹脂:活性基團為叔胺基團 弱堿性陰離子交換樹脂:活性基團為伯胺或仲胺 R-N+(CH3)3OH- +Cl-
—— R-N+(CH3)3Cl-+OH
-
疏水性:
纖維素離子交換劑:分陽離子與陰離子交換纖維,骨架松散、親水性強、表面積大、交換容量大、吸附力弱、交換和洗脫條件溫和、分辨率高。
常用的有:甲基磺酸纖維素、羧甲基纖維素(CMC)、二乙基氨基乙基纖維素(DEAE)
交聯葡聚糖離子交換劑:分陽離子與陰離子交換樹脂,除了具有離子交換功能以外,兼有分子篩的功能,提高了分離的效率。
常用:CM-sephadex C-25、DEAE-sephadex A-50等。
瓊脂糖離子離交換劑:將電荷基團(DEAE-或CM-等基團)附著在Sepharose CL-6B 上形成
常用:DEAE-Sepharose(陰離子)、CM-Sepharose(陽離子)
2. 洗脫順序:平衡離子反電荷物質/中性物質、小結合力物質、大結合力物質。
3. 結合強度的影響因素:離子交換劑性質、離子自身性質、離子強度、pH、溫度、溶劑組成等。
eg.蛋白等生物大分子通常呈兩性,與pH關系較大,在一定的pH條件下,等電點pI高于pH的蛋白帶正電,能與陽離子交換劑結合,一般pI越高的蛋白與離子交換劑結合力越強。
4. 交換容量的測定:陽離子交換劑shou先用HCl處理,使其平衡離子為H+,再用水洗**中性,對于強酸型離子交換劑,用NaCl充分置換出H+,再用標準濃度的NaOH滴定生成的HCl,就可以計算出離子交換劑的交換容量;對于弱酸型離子交換劑,用一定量的堿將H+充分置換出來,再用酸滴定,計算出離子交換劑消耗的堿量,就可以算出交換容量。陰離子交換劑方法同此。 5. 離子交換劑的選擇:
陰陽選:被分離物質帶正電,選陽離子交換劑;被分離物質帶負電,選陰離子交換劑。 #p#分頁標題#e#
緩沖液pH比有效成分的pI高一個單位(選用陰離子交換劑) 緩沖液pH比有效成分的pI低一個單位(選用陽離子交換劑)
基團選:小分子物質/極端pH條件分離,選強酸或強堿型離子交換劑;蛋白質等大分子易變性,選弱酸或弱堿型離子交換劑。
基質選:聚苯乙烯離子交換劑(聚苯乙烯樹脂),機械強度大、流速快、強疏水性、易使蛋白變性,常用于分離小分子物質如無機離子、氨基酸、核甘酸;纖維素(Cellulose)、球狀纖維素(Sephacel)、葡聚糖(Sephadex)、瓊脂糖(Sepharose),與水有較強親和力,適合蛋白質等大分子物質。[纖維素,價格低,分辨率、穩定性低,適于初步分離與大量制備;葡聚糖,分辨率、價格適中,受外界影響大(體積受離子強度與pH影響大);瓊脂糖,分辨率、穩定性高,價格貴。]
eg. DEAE-纖維素(二乙基氨基纖維素)和CM-纖維素(羧甲基纖維素)交換容量大、親水性、洗脫溫和、回收率高,在生物大分子物質(蛋白質,酶,核酸等)的分離中**常用。
顆粒選:顆粒小,分辨率高,平衡時間長,流速慢,適合于對分辨率要求不高,大規模制備性分離;顆粒大,分辨率低,平衡時間短,流速快,適合于對分辨率要求較高,小規模分析性分離。 6. 離子交換劑的處理保存:
處理程序:
膨化,干粉在水中溶脹,用水懸浮,除雜質與細小顆粒;
新出廠干樹脂用水浸泡2小時后減抽壓去氣泡,傾去水,再用大量無離子水洗**澄清,去水后加4倍量2M HCl攪抖4小時,除去酸液,水洗到中性,再加4倍量2M NaOH攪拌4小時,除堿液,水洗到中性備用。
酸堿浸泡,原則濃度不宜過高、時間不宜過長、溫度不宜過高,每種溶劑浸泡間用水洗**中性,陽離子交換劑后用堿處理(NaOH+NaCl,Na型),陰離子交換劑后用酸處理(HCl,Cl型),一般濃度低于0.5mol/L,浸泡時間30min,**后也用水洗**中性(為進一步除雜,帶平衡離子,通常陽離子交換劑帶Na離子,陰離子交換劑帶Cl離子),除盡氣泡; 再生,酸堿交替浸泡,選擇合適的平衡離子,同前。 保存,洗凈,加防腐劑(0.02%疊氮鈉),4℃下保存,也可保存在10mM NaOH/20%乙醇溶液或是兩者的混合液中。
7. 層析基本操作流程:
裝柱,粗而短,不宜過長過短,垂直,均勻,平整,無氣泡; 前處理,(再生的一個過程)高濃度堿鹽(1M NaCl,1~2個柱床體積)將層析介質原掛有的離子置換下,使平衡過程中能被置換的離子均勻掛上;(少用) 平衡,平衡體系一般采用緩沖溶液+保護劑的模式,常用的平衡體系有TE緩沖體系、Ac緩沖體系、PB緩沖體系和碳酸緩沖體系等,常用的保護劑有EDTA及吐溫系列等(為了達到較好的平衡效果,在平衡前經常加一預平衡過程,采用的是較高濃度的緩沖體系沖一柱床體積,目的是為了較快的將層析柱上的酸堿離子置換下來。);
上樣,檢測樣品液離子強度與pH值,應與緩沖體系pH值相當(±0.5); 沖柱,平衡緩沖液,1~2個柱體積;
洗脫,梯度(改變離子強度或pH,改變離子強度通常是在洗脫過程中逐步增大離子強度,從而使與離子交換劑結合的各個組分被洗脫下來;而改變pH的洗脫,對于陽離子交換劑一般是pH從低到高洗脫,陰離子交換劑一般是pH從高到低),流速恒定(提高分辨可通過降低流速); 樣品收集,一般收集3/5/6個樣品; 后處理,同前處理;
封柱,若短時間不用,則用封柱液(封柱液有10mM的NaOH、20%乙醇和0.1%的疊氮鈉)封柱。 8. 實例
水處理:一般是將水依次通過H+型強陽離子交換劑,去除各種陽離子及與陽離子交換劑吸附的雜質;再通過OH- 型強陰離子交換劑,去除各種陰離子及與陰離子交換劑吸附的雜質,即可得到純水。再通過弱型陽離子和陰離子交換劑進一步純化,就可以得到純度較高的純水。
小分子氨基酸:使用強酸性陽離子聚苯乙烯樹脂,將氨基酸混合液在pH 2~3上柱。這時氨基酸都結合在樹脂上,再逐步提高洗脫液的的離子強度和pH,這樣各種氨基酸將以不同的速度被洗脫下來,可以進行分離鑒定。
親和層析
1. 本質:靜電作用、氫鍵、疏水作用、配位鍵、弱共價鍵…… 2. 專一親和配對物:
抗原與抗體、DNA與互補DNA或RNA、酶與它的底物或競爭性抑制劑、激素(或藥物)與它們的受體、維生素和它的特異結合蛋白、糖蛋白與它相應的植物凝集素等。
eg.蛋白質-組氨酸
3. 影響因素:
離子強度、pH值、氫鍵抑制劑(脲、鹽酸胍)、溫度(提高溫度,靜電作用、氫鍵、配位鍵減弱;但是,疏水性相互作用增強)、螯合劑(影響配位鍵)、離液離子(SCN-、I-、CIO4-,疏水作用減弱)。 4. 過程:載體活化、配基連接、吸附、吸脫。 5. 介質:基質-手臂-配體。
基質:纖維素、聚丙烯酰胺凝膠、交聯葡聚糖、瓊脂糖、交聯瓊脂糖、多孔玻璃珠等。
eg. Sepharose 4B
配體:特異性配體。eg. 生物素和親和素、抗原和抗體、酶和它的抑制劑、激素-受體等。 通用性配體。eg. 凝集素。 #p#分頁標題#e#
手臂:適當長度的氨基化合物NH2(CH2)nR(R=氨基/羧基,n=2~12) 6. 制備過程:
基質活化:
溴化腈活化法,方法,瓊脂糖與等體積水混合,加入溴化腈,用NaOH調節pH11,20℃維持3~12 min,迅速加入大量冰屑,布氏漏斗抽濾,緩沖液抽濾洗滌,反應過程如下:
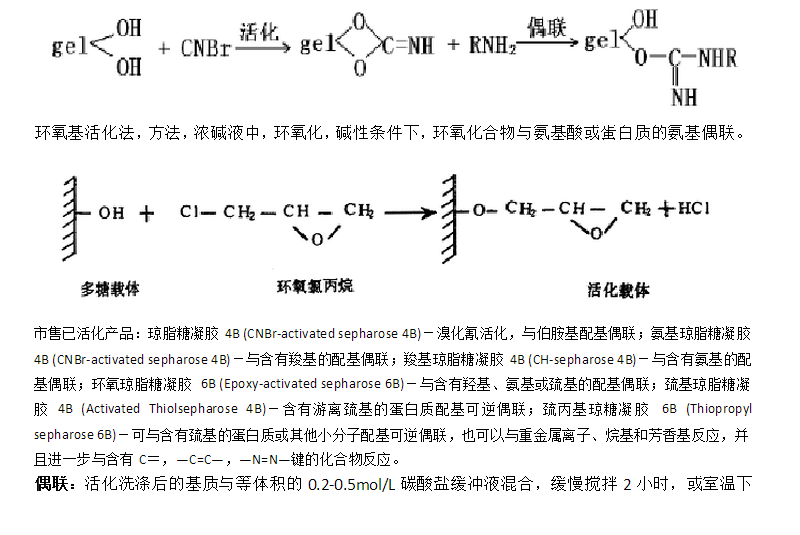
過夜,以便配體和基質充分偶聯而形成固體載體,對于偶聯極牢固會喪失活性的大分子物質配體,則應在低pH值緩沖液中反應或活化基質預先在pH8.3的堿性溶液中進行適當的水解作用,這樣可提高配體的活性,配體和基質偶聯完畢后,必須要反復洗滌,以去除未偶聯的配體,另外要用適當的方法封閉基質中未偶聯上配體的活性基團,也就是使基質失活,以免影響后面的親和層析分離。
eg.對于能結合氨基的活性基團,常用的方法是用2-乙醇胺、氨基乙烷等小分子處理。
配體結合量測定:測定方法有直接測定法(2,4,6-三硝基苯磺酸鈉的顏色試驗和根據配體的特征來進行測定)和間接測定法(推算法和根據親和吸附劑對被吸附物質的操作容量可計算配體的結合量)。 7. 分離過程:在低溫下進行(4℃)
平衡:2~3倍體積緩沖液。
吸附:選擇吸附柱時,吸附能力強用短柱,吸附能力弱時用長柱;上樣溶劑用起始緩沖液,可用起始緩沖液溶解固體,已溶溶液用透析將溶劑換成起始緩沖液;上樣量小于吸附容量1/3,吸附力弱則為1/10。
沖洗:10倍柱體積緩沖液洗掉不結合的雜質,流速為獲得**尖銳洗脫峰和**小洗脫體積的流速。 洗脫:洗脫時可先讓洗脫劑在柱子中停留半小時的方法,盡量避免分離物質失活。(洗脫后應注意中和酸堿,透析去除離子,以免待分離物質喪失活性。) 保存:使用過的親和層析柱,一般用大量的洗脫液或較高濃度的鹽溶液洗滌,**后用平衡緩沖液(平衡緩沖液應與樣品緩沖液一致)使親和層析柱平衡,即可重復使用,暫時不用時,加入0.01%的疊氮化鈉,4℃下保存,也可以加入0.5%的醋酸洗必泰或0.05%的苯甲酸。應注意不要使親和吸附劑冰凍。(注意處理時不能改變配體的活性。) 8. 應用:
純化大分子物質:抗原、抗體,糖蛋白,核酸等稀溶液的濃縮;不穩定蛋白質的貯藏;從純化的分子中除去殘余的污染物;用免疫吸附劑吸附純化對此尚無互補配體的生物大分子;分離核酸是親和層析應用的一個重要方面。
研究酶的結構與功能:使用專一的化學試劑改變酶的功能團,會引起酶活力的不完全喪失,難以確定酶活力的改變是化學試劑的作用還是殘留的天然酶活力,利用親和層析能有效地將失活的酶和天然的酶有效的分離
親和色譜可用于下列生物體系
酶:底物、抑制劑、輔酶;抗體:抗原、病毒、細胞;外源凝集素:多糖、糖蛋白、細胞表面受體;細胞核酸:互補堿基序列、組蛋白、核酸聚合酶 、結合蛋白;激素及維生素:受體、載體蛋白;細胞:細胞表面特異蛋白、外源凝集素
其他層析
1. 吸附層析 2. 聚焦層析 3. 疏水層析