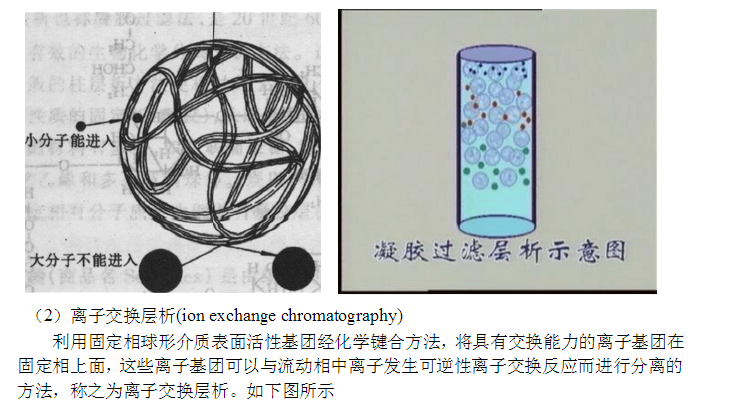
利用凝膠層析介質(固定相)交聯度的不同所形成的網狀孔徑的大小,在層析時能阻止比網孔直徑大的生物大分子通過。利用流動相中溶質的分子量大小差異而進行分離的一種方法,稱之為排阻層析。如下圖所示
利用固定相載體上偶聯的亞胺基乙二酸為配基與二價金屬離子發生螯合作用,結合在固定相上,二價金屬離子可以與流動相中含有的半胱氨酸、組氨酸、咪唑及其類似物發生特異螯合作用而進行分離的方法,稱之為金屬螯合層析。如下圖所示
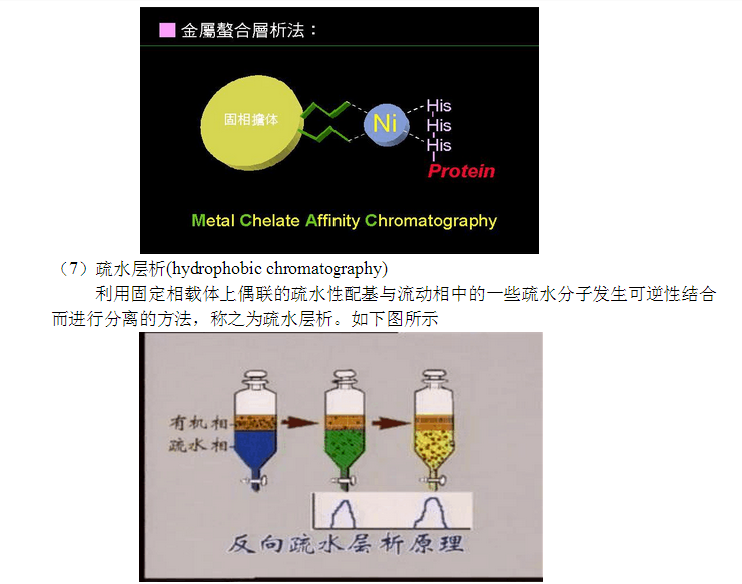
(8)反向層析(reverse phase chromatography)
利用固定相載體上偶聯的疏水性較強的配基,在一定非極性的溶劑中能夠與溶劑中的疏水分子發生作用,以非極性配基為固定相,極性溶劑為流動相來分離不同極性的物質的方法,稱之為反相層析。
(9)聚焦層析(focusing chromatography)
利用固定相載體上偶聯的載體兩性電解質分子,在層析過程中所形成的pH梯度,并與流動相中不同等電點的分子發生聚焦反應進行分離的方法,稱之為聚焦層析。 (10)灌注層析(perfusion chromatography)
利用剛性較強的層析介質顆粒中具有的不同大小貫穿孔與流動相中溶質分子分子量的差異進行分離的方法,稱之為灌注層析。 2. 常用層析技術
2.1 離子交換層析技術
是以離子交換纖維素或以離子交換葡聚糖凝膠為固定相,以蛋白質等樣品為移動相,分離和提純蛋白質、核酸、酶、激素和多糖等的一項技術。 2.1.1 原理
在纖維素與葡聚糖分子上結合有一定的離子基團,當結合陽離子基團時,可換出陰離子,則稱為陰離子交換劑。如二乙氨乙基(Dicthylaminoethyl,DEAE)纖維素。在纖維素上結合了DEAE,含有帶正電荷的陽離子纖維素—O—C6 H14N+H,它的反離子為陰離子(如Cl-等),可與帶負電荷的蛋白質陰離子進行交換。當結合陰離子基團時,可置換陽離子,稱為陽離子交換劑,如羧甲基(Carboxymethy, CM)纖維素。纖維素分子上帶有負電荷的陰離
子(纖維素-O-CH2-COO一
),其反離子為陽離子(如Na+等),可與帶正電荷蛋白質陽離子進行交換。
溶液的pH值與蛋白質等電點相同時,靜電荷為0,當溶液pH值大于蛋白質等電點時,則羧基游離,蛋白質帶負電荷。反之,溶液的pH值小于蛋白質等電點時,則氨基電離,蛋白質帶正電荷。溶液的pH值距蛋白質等電點越遠,蛋白質的電荷越多。反之則越少。血清蛋白質均帶負電荷,但各種蛋白質帶負電荷的程度有所差異,以白蛋白為**多,依次為?球 蛋白,β球蛋白和γ球蛋白
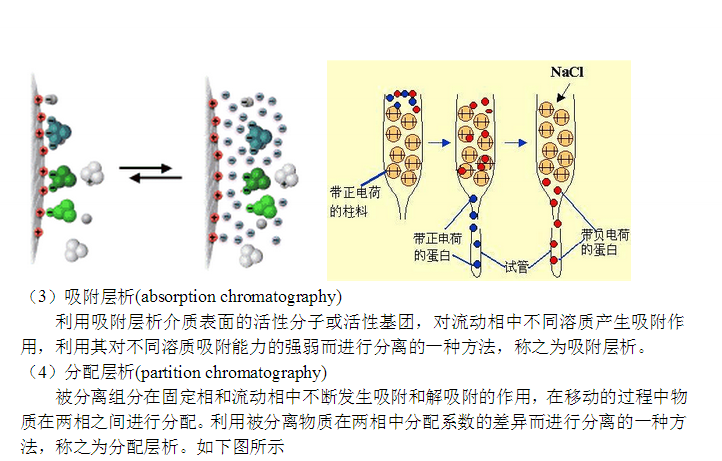
在適當的鹽濃度下,溶液的pH值高于等電點時,蛋白質被陰離子交換劑所吸附;當溶液的pH值低于等電點時,蛋白質被陽離子交換劑所吸附。由于各種蛋白質所帶的電荷不同。它們與交換劑的結合程度也不同,只要溶液pH值發生改變,就會直接影響到蛋白質與交換劑的吸附,從而可能把不同的蛋白質逐個分離開來。
交換劑對膠體離子(如蛋白質)和無機鹽離子(如NaCl)都具有交換吸附的能力,當#p#分頁標題#e#
兩者同時存在于一個層析過程中,則產生競爭性的交換吸附。當Cl一
的濃度大時,蛋白質不
容易被吸附,吸附后也易于被洗脫,當Cl一
濃度小時,蛋白質易被吸附,吸附后也不容易被洗脫。因此,在離子交換層析中,一般采用兩種方法達到分離蛋白質的目的。一種是增加洗脫液的離子強度,一種是改變洗脫液的pH值。pH值增高時,抑制蛋白質陽離子化,隨之對陽離子交換劑的吸附力減弱。pH值降低時,抑制蛋白質陰離子化,隨之降低了蛋白質對陰離子交換劑的吸附。當使用陰離子交換劑時,增加鹽離子,則降低pH值。當使用陽離子交換劑時,增加鹽離子濃度,則升高溶液pH值。 2.1.2常用離子交換劑的種類與特性
(1)離子交換纖維素 離子交換纖維素的種類很多,**常用的是DEAE—纖維素和CM纖維素。由于劑型不同,其理化性質和作用也有所差異。一般而言,微粒型要優于纖維素型,因為微粒型是在纖維素型的基礎上進一步提煉而成。它的交換容量大,粒細、比重大,能裝成緊密的層析柱,要求分辨力高的實驗可用此型纖維素。
離子交換纖維素的優點為:①離子交換纖維素為開放性長鏈,具有較大的表面積,吸附容量**大;②離子基團少,排列稀疏,與蛋白質結合不太牢固,易于洗脫;③具有良好的穩定性,洗脫劑的選擇范圍廣。
(2)離子交換交聯葡聚糖 離子交換交聯葡聚糖也是廣泛使用的離子交換劑,它與離子交換纖維素不同點是載體不同。
離子交換交聯葡聚糖有如下優點:①不會引起被分離物質的變性或失活;②非特異性吸附少;③交換容量大。
離子交換葡聚糖的選用,一般根據蛋白質的分子量而定。中等分子量(30 000-200 000)一般選A50和C50,而低分子量(<30 000和高分子量>200 000)均宜選用A25和C25。 2.1.3 試驗方法
陰離子交換劑與陽離子交換劑的裝柱和層析過程基本相同。交聯葡聚糖的預處理只需充分溶脹和平衡,不需要除去細粒碎片和酸堿處理。其他步驟也基本同離子交換纖維素。
(1)劑型的選擇 根據蛋白質在所用緩沖液pH值下帶電荷的種類選擇,如pH高于蛋白質等電點,應選陰離子交換劑,反之應選陽離子交換劑。一般情況下,DEAE-纖維素用于分離酸性蛋白,而CM纖維素用于分離堿性蛋白質。 下面以DEAE-纖維素操作為例,介紹試驗方法
(2)膨脹活化 此步的目的在于除去雜質,暴露DEAE-纖維素上的極性基團。DEAE-纖維素的用量則根據柱容積的大小和所需過柱樣品的量來決定。一般是1.0g DEAE-纖維素相當于6ml~8ml柱床體積。
稱取所需的量,撒于0.5 mol/L NaOH溶液中(1g DEAE—纖維素干粉約需15倍NaOH液),浸泡1h左右,不時攪拌。抽濾(以布氏漏斗加兩層濾紙或尼龍紗布抽濾),以蒸餾水洗滌,再抽濾,直**濾液近中性為止,再將纖維素浸泡于0.5Mol/L HCl中1h,同樣抽濾液**近中性。再將纖維素浸于0.5 mol/L NaOH液中,同樣處理,洗**中性。
(3)平衡 將DEAE—纖維素放入0.0l mol/L pH 7. 4 PB液中(即起始緩沖液),靜止1 h,不時攪拌,待纖維素下沉后,傾去上清液或抽濾除去洗液,如此反復幾次**傾出液體的pH值與加入的PB液的pH值相近時為止。
(4)裝柱 層析柱的選擇要大小、長度適當。一般而言,柱長和柱直徑之比為10︰1~20︰1,柱的內徑上下要均勻一致。用前將層析柱在清潔液內浸泡處理24 h,然后依次用常水、蒸餾水、起始緩沖液充分洗滌。
(5)上樣 要層析的樣品shou先必須用起始緩沖液(4℃)平衡過夜,中間可換液數次。將柱的上端打開,用吸管將纖維素柱上面的緩沖液吸出,不要吸凈,留一薄層液面,以免空氣進入。沿管壁緩緩加入樣品,注意不要打亂纖維素表層。擰開下端的螺旋夾,使樣品進入交換劑中,快要進完時,加1 ml~2 ml緩沖液沖洗柱壁,隨即用多量的洗脫液洗脫。
樣品的加量與DEAE—纖維素有一個**適比的關系,超過這個比值,吸附就不完全,直接影響到分離的純度。經過粗提的?—球蛋白50 mg~100 mg,用干重約4 g DEAE-纖維素裝柱分離,可獲得理想結果。
(6)洗脫 對于陰離子交換劑而言,洗脫的辦法是使pH逐漸降低,而離子濃度逐漸升高。一般的辦法,是穩定一個因素而改變另一個因素洗脫。洗脫可采用分段洗脫和連續洗脫法,前者較實用,后者較準確。
(7)洗脫液的收集 利用自動分步收集器收集,并以20%磺基水楊酸測試,當蛋白液下來時,開始分管收集,**無蛋白液為止。
(8)交換柱的再生 將使用過的DEAE-纖維素移入燒杯中,用2 mol/L NaCl液浸泡,抽濾并洗滌數次。如不立即使用,可加1/10 000的疊氮鈉防腐,保存于4℃冰箱中。使用時,再以堿-酸-堿處理。
2.2 凝膠層析
凝膠層析又稱為分子篩層析或凝膠過濾。具有分子篩作用的物質很多,如浮石、瓊脂、瓊脂糖、聚乙烯醇、聚丙烯酰胺、葡聚糖凝膠等。以葡聚糖凝膠應用**廣,商品名是sephadex型號很多,從G10到G200,它的主要應用范圍是:①分級分離各種抗原與抗體;②去掉復合物中的小分子物質。如除鹽、熒光素和游離的放射性同位素以及水解的蛋白質碎片;③分析血清中的免疫復合物;④分子量的測定。 2.2.1 原理 #p#分頁標題#e#
凝膠是一種多孔性的不帶表面電荷的物質,當帶有多種成分的樣品溶液在凝膠內運動時,由于它們的分子量不同而表現出速度的快慢,在緩沖液洗脫時,分子量大的物質不能進入凝膠孔內,而在凝膠間幾乎是垂直的向下運動,而分子量小的物質則進入凝膠孔內進行“繞道”運行,這樣就可以按分子量的大小,先后流出凝膠柱,達到分離的目的。 2.2.2 葡聚糖凝膠的種類與性能 葡聚糖又名右旋糖酐,在它們的長鏈間以三氯環氧丙烷交聯劑交聯而成。葡聚糖凝膠具有很強的吸水性,交聯度大,吸水性小,相反交聯度小,吸水性大。商品名以SephadexG表示,G值越小,交聯度越大,吸水性越小,G值越大,交聯度越小,吸水性就越大,二者呈反比關系,G值大約為吸水量的10倍。由此可以根據床體積而估算出葡聚糖凝膠干粉的用量。
G25、G50有四種顆粒型號:粗(100µ~300µ)、中(50µ~150µ)、細(20µ~80µ)和超細(10µ~40µ)。G75~G200又有兩種顆粒型號:中(40µ~120µ),超細(10µ~40µ)。顆粒越細,流速越慢,分離效果越好。 2.2.3 試驗方法
(1)凝膠的選擇 根據層析物質分子量的大小選擇不同型號的凝膠,如除鹽和除游離的熒光素,則可選用粗、中粒度的G28或G500,G250多用于分離蛋白質單體,G200多用于分離蛋白質凝膠聚合體等。
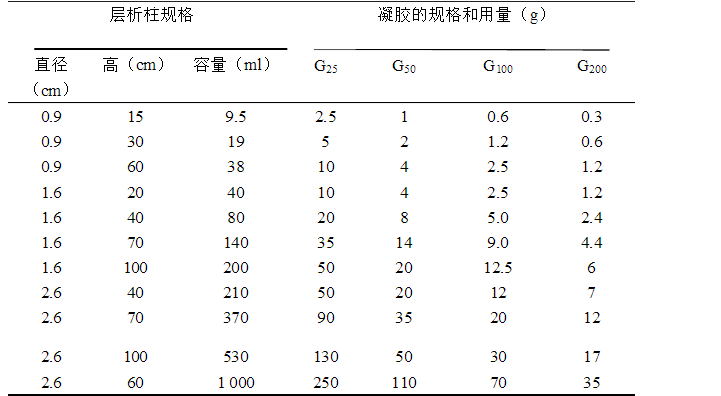
(2)凝膠的預處理 稱取適量的凝膠加入過量的緩沖液在冰箱(或室溫)中充分膨脹,或在沸水中煮,膨脹時間應根據不同型號的凝膠而定。如下表所示
凝膠量與型號和層析柱大小與規格及凝膠用量
為使粒子均勻一致需進行浮選,即加入凝膠粒子后,輕輕攪拌,靜置20 min,傾去沉淀的粒子,如此反復數次即可。
(3)裝柱 層析柱的選擇一般根據分離樣品的種類和樣品的數量而定。純化蛋白質時,柱床體積應為樣品體積的25~100倍。去鹽、游離熒光素約為樣品體積的4~10倍。柱太短,影響分離效果。柱長一些,分離效果好,但柱太長,則延長分離時間,樣品也稀釋過度。層析柱的內徑也要選擇適當。內徑過細,會發生“器壁效應”,即靠近管壁的流速要大于中心的流速影響分離效果。所以層析柱的內徑和高度應有一定的比例。對于除鹽來說應為1︰5~1︰25;對于純化蛋白質來說應為1︰20~1︰100。 裝柱過程基本同離子交換層析柱。
(4)加樣與洗脫 樣品體積不宜過多,**好為床體積的1%~5%,**多不要超過10%。樣品濃度也不宜過大,濃度過大粘度大,分離效果差,一般不超過4%。
洗脫液應與膨脹一致,否則更換溶劑,凝膠體積會發生變化,影響分離效果。洗脫液要有一定的離子強度和pH值。分離血清蛋白常用0.02~0.1 mol/L pH 6.9~8.0的PBS液(0.14 mol/L NaCl)和0.1 mol/L pH8.0 Tris-HCl緩沖鹽溶液(0.14 mol/L NaCl)。
(5)洗脫液收集 同離子交換層析。
(6)凝膠柱的重復使用與保存 當樣品的各組分全部洗脫下來之后,即可加入新的樣品,繼續使用。保存方法有三種:
A 在液相中保存**方便,即于凝膠懸液中加入防腐劑(一般為0.02% N2N3或0.002%洗必泰)或高壓滅菌后4℃保存。此法**少可以保存半年以上。
B 用完后,以水沖洗,然后用60%~70%酒精液沖洗,凝膠體積縮小,即在半收縮狀態下保存。
C 長期不用者,**好以干燥狀態保存,即水洗凈后,用含乙醇的水洗,逐漸加大乙醇用量,**后用95%的乙醇洗,可全部去水,再用乙烯去除乙醇,抽濾干,于60℃~80℃干燥后保存。
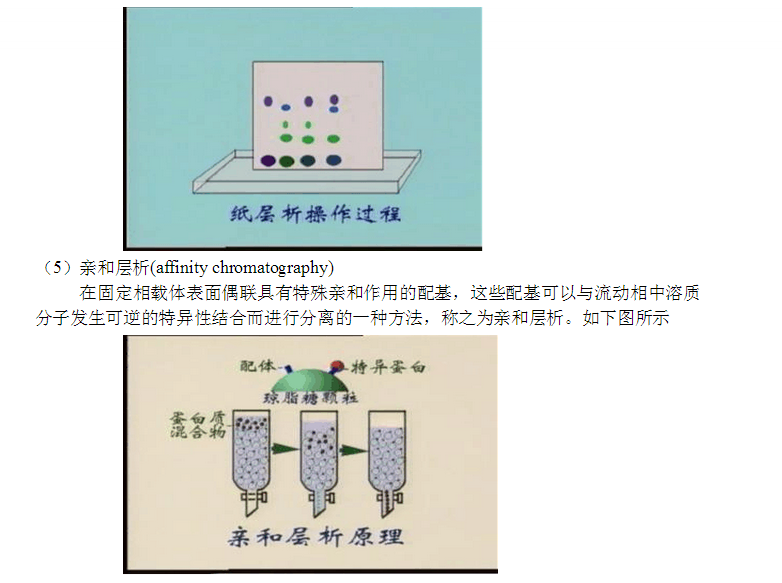
2.3 親和層析 2.3.1 原理
親和層析是一種吸附層析,抗原(或抗體)和相應的抗體(或抗原)發生特異性結合,而這種結合在一定的條件下又是可逆的。所以將抗原(或抗體)固相化后,就可以使存在液相中的相應抗體(或抗原)選擇性地結合在固相載體上,借以與液相中的其他蛋白質分開,達到分離提純的目的。
此法具有高效、快速、簡便等優點。 2.3.2載體的基本要求和選擇
理想的載體應具有下列基本條件:①不溶于水,但高度親水;②惰性物質,非特異性吸附少;③具有相當量的化學基團可供活化;④理化性質穩定;⑤機械性能好,具有一定的顆粒形式以保持一定的流速;⑥通透性好,**好為多孔的網狀結構,使大分子能自由通過;⑦能抵抗微生物和醇的作用。
可以做為固相載體的有皂土、玻璃微球、石英微球、羥磷酸鈣、氧化鋁、聚丙烯酰胺凝膠、淀粉凝膠、葡聚糖凝膠、纖維素和瓊脂糖。在這些載體中,皂土、玻璃微球等吸附能力弱,且不能防止非特異性吸附。纖維素的非特異性吸附強。聚丙稀酰胺凝膠是目前的**優良載體。
瓊脂糖凝膠的優點是親水性強,理化性質穩定,不受細菌和酶的作用,具有疏松的網狀結構,在緩沖液離子濃度大于0.05 mol/L時,對蛋白質幾乎沒有非特異性吸附。瓊脂糖凝膠極易被溴化氫活化,活化后性質穩定,能經受層析的各種條件,如0.1 mol/L NaOH或1 mol/L HCl處理2 h~3 h及蛋白質變性劑7 mol/L尿素或6 mol/L鹽酸胍處理,不引起性質改變,故易于再生和反復使用。 #p#分頁標題#e#
瓊脂糖凝膠微球的商品名為Sepharose,含糖濃度為2%、4%、6%時分別稱為2B、4B、6B。因為Sepharose 4B的結構比6B疏松,而吸附容量比2B大,所以4B應用**廣。 2.3.3 試劑與配制 1.Sepharose 4B 2.CNBr(劇毒) 3.抗原或抗體
4.1 mol/L NaHCO3 取NaHCO3 84.01g加水**1 000 ml。 5.0.1 mol/L NaHCO3 6.2 mol/L NaOH
7.0.01 mol/L pH7.4 PBS
0.2 mol/L Na2HPO4 38.0 ml 0.2 mol/L Na2HPO4 162.0 ml NaCl 32.76 g 加水**4 000 ml。 8.0.1 mol/L pH 2.8 甘氨酸即氨基乙酸—HCl緩沖液 甘氨酸15.01 g加水**1 000 ml,取此液,加0.2 mol/L HCl 84 ml加水166 ml共500 ml。 9.7 mol/L尿素
10.0.2 mol/L NaHCO3,(含0.1 mol/L NaCI) 1 mol/L NaHCO3 100.00 ml NaCl 2.93 g 加水**500.00 ml。 2.3.4 實驗方法 1.瓊脂糖活化 ⑴ 取20 ml Sepharose 4B放在布氏漏斗中抽干,加少量的0.1 mol/L pH 9.0 NaHCO3液洗滌,立即轉入100 ml燒杯中,冰浴置于磁力攪拌器上。 ⑵ 在通風櫥內稱取2 g溴化氰,加水20 ml溶解,然后倒入瓊脂糖中,小心滴加2 mol/L NaOH,使pH保持在11左右,反應10 min。在1 min~2 min迅速調整pH為8.0~11.0維持10 min。 ⑶ 將活化的瓊脂糖迅速倒入布氏漏斗中,以冰水抽洗成中性,再迅速以250 ml冷的0.1 mol/L pH 9.0 NaHCO3抽洗。
2.偶聯蛋白 20 ml 4B液體相當于一半的固相載體。 ⑴ 事先將需偶聯的抗原(或抗體)蛋白200 mg置于0.1 mol/L pH 9.0 NaHCO3液中透析數小時(一般偶聯量為10~30 mg/g載體)。
⑵ 將活化的瓊脂糖迅速倒入蛋白液中(在1.5 min內,從抽洗到偶聯),4℃緩慢攪拌過夜,使蛋白與活化的瓊脂結合。
3.裝柱 ⑴ 選柱:不宜過大,一般以1.5×15 cm的層析柱可裝偶聯蛋白的瓊脂糖約30ml。 ⑵ 裝柱:將已與蛋白偶聯的瓊脂糖裝入層析柱內,擰緊下口夾,讓其下沉,數分鐘后松開下口夾,使溶液約以1 ml/min的速度流出。 ⑶ 洗柱:以0.2 mol/L pH 9.0 NaHCO3(含0.1 mol/L NaCl)洗滌**洗出液的OD280<0.02為止。
收集全部的洗脫液,測得的OD280值×洗脫液的總ml數即為未偶聯蛋白的含量,由此可計算偶聯率。


 #p#分頁標題#e#
#p#分頁標題#e#


